Session 4: Running a project
Here you will learn several concepts needed for running real projects:
use one screen session per project (if you’re juggling more projects)
keep your code in a text file (using
nanotext editor)keep your data files separate from your code
make your intent more legible by wrapping code in functions and script files
write handy scripts in
awkdo the same thing with many files at once
speed up your processing by parallelizing the work
Keep your stuff ordered
Let’s pretend we’re starting to work on something serious, a new project:
cd ~/projects
# a new project dir, separate dir for data in one shot
mkdir -p unix-advanced/data
# set your work dir to the 'current' project dir
cd unix-advanced
# definitely run screen to be safe
# screen sessions can be named, which helps you
screen -S advanced
# prepare source data
cp /data-shared/bed_examples/Ensembl.NCBIM37.67.bed data/
# add a new window with ctrl+a c
# note and save all the (working) code
nano workflow.sh
Note
The nano bottom bar has a lot of weird symbols. ^O means type ctrl-o,
pressing both of the keys at once. M-U means alt-u, again pressing both
simultaneously. For some reason, Write Out means Save in usual parlance.
To edit a file on the remote machine, you have to use a text editor on that machine.
One of the most basic editors is nano.
You’ll be using screen to keep two windows open at once. In the first window, you’ll be experimenting with shell commands. In the second one, you’ll keep track of the final versions of your commands, that comprise your workflow.
That is - after you’re satisfied with your command at the bash prompt, copy it to
the second window where you have nano open, and hit ctrl-o to save it.
You’ll be writing code on your own now, so you need to keep track of what you created. I’ll occasionally post solutions to Slack.
awk (pronounced [auk])
awk is most often used instead of cut, when the fields are separated
by spaces (not tabulators) and padded to a fixed width. awk can ignore the whitespace.
awk also allows you to reorder the columns. cut can’t do this:
But what if you wanted to have a table which mentions chromosomes first and
then the counts? Enter awk. Every line (called record) is split
into fields, which are assigned to variables $1 for first field,
$2 for second etc. The whole line is in $0 if needed. awk expects
the program as first argument:
Additionally you can add conditions when the code is executed:
Or even leave out the code body, which is the same as one {print $0}
statement - that is print the matching lines:
There are some other variables pre-filled for each line, like
record number NR (starting at 1) and number of fields NF.
# NF comes handy when checking if it's okay to
# process a file with (say) cut
<$INPUT awk '{print NF}' | uniq
Let’s play with some fastq files. Extract first five files to data.
We’ll explain how xargs works later:
INPUT=/data-shared/fastq/fastq.tar.gz
<$INPUT tar tz | head -5 | xargs tar xvf $INPUT -C data
Look at the data with less - these are reads from 454, with varying read lengths.
Let’s check the lengths:
<data/HRTMUOC01.RL12.01.fastq paste - - - - | awk '{print $1, length($2)}' | head
What paste does here is it takes four lines from the input and puts them side by side,
separated by \t characters (and repeats for the rest of the input).
Let’s filter on the sequence length, keeping only reads longer than 100 bases.
We’d like to output a valid fastq file (that means reversing the paste operation):
What have we learned?
use
awkwhencutfalls shortuse
awkto filter rows based on field valuesuse
awkto create summary tables
Functions in the Shell
This creates a command called uniqt that will behave as uniq -c, but
there will be no padding (spaces) in front of the numbers, and numbers will be
separated by <tab>, so you can use it with cut.
uniqt() { uniq -c | sed -r 's/^ *([0-9]+) /\1\t/' ;}
Now test it:
<data/Ensembl.NCBIM37.67.bed cut -f1 | sort | uniqt | head
You can see that the basics of the syntax are your-name() { command pipeline ;}.
If you want to pass some arguments into the function, use $1, $2 etc.:
test-function() { echo First argument: $1 ;}
test-function my-argument
When not given any inputs, the command behaves in the ‘unix way’ - it reads from stdin and writes to stdout.
Now create a function called fastq-min-length, with one argument
(use $1 in the body of the function) giving the minimal length:
There is a problem with quoting. Awk uses $1 for something else than the
shell, we need to protect $1 with single quotes, but we still need to access
shell’s $1 somehow… Awk’s -v argument helps in this case - use
it like awk -v min_len=$1 '(length($2) > min_len)'.
Note
Let’s pop-open the matryoshka. What is terminal, what is a shell, what is Bash?
The program which takes care of collecting your keystrokes and rendering
the colored characters which come from the server is called a terminal.
Famous terminals are mintty (that’s what you’re using in Windows now),
Konsole, Terminal App… The next doll inside is ssh. It takes
care of encrypted communication with the remote server. An interesting
alternative for geeks is mosh (google it yourself;). Now you need a
program to talk to on the remote side - that is the shell. We’re using
bash now, sometimes you can meet the simpler cousin sh, and the kool
kids are doing zsh. To recap, Bash is to shell what Firefox is to
browser.
What have we learned?
use
function_name() { your_code_here ;}to create a functionuse
$1,$2etc. to access arguments you pass on the command lineuse
-vto pass arguments toawkto resolve quoting issues
Shell Scripts
Another way to organize your code is to put it into a separate file
called a ‘script file’. It begins with a shebang line, telling the computer
which language is the script in. Bash shebang is #! /bin/bash.
Take care to give a descriptive name to your script:
nano fastq-filter-length.sh
Copy and paste the following code block into the nano editor, save it with ctrl+o
and switch to another bash window in screen.
We need to mark the file as executable and test it:
chmod +x fastq-filter-length.sh
# check with ls, filter_fastq.sh should be green now
# and using ll you should see the 'x' (eXecutable) permission
ls
ll
# and run it (the ./ is important!)
./fastq-filter-length.sh
Note
You can check file permissions by typing ll instead of ls.
rwx stand for Read, Write, eXecute, and are repeated three times,
for User, Group, and Others. The two names you see next to the
permissions are file’s owner user and group.
You can change the permissions - if you have the permission to do so -
by e.g. chmod go+w - “add write permission to group and others”.
Now collect your code from above (contents of your function, not the whole
function) and paste it below the shebang. Don’t forget to remove the debug echo
parts - otherwise your script will spoil it’s output with some useless chatter.
# when the final code is there, you need to give it input (and maybe save the output):
<data/HRTMUOC01.RL12.01.fastq ./fastq-filter-length.sh 90 > data/filtered.fastq
What have we learned?
save your code in a reusable file that you can run from the command line
use shebang line and
chmod +xto make your script executableuse
$1,$2etc. to access arguments you pass on the command line
Process multiple files
Multi-file processing is best done with find and xargs.
Let’s check the basic concepts - find converts directory structure to
‘data’ (stdout), xargs converts stdin to command line(s).
# Investigate!
find data -type f
# here xargs appends all the lines from stdin to the command
find data -type f | xargs echo
# here xargs replaces the {} with the line from stdin
# and when there are more lines, it runs the command multiple times
find data -type f | xargs -I{} echo File: {} found!
There is a lot of filtering options built-in to find. You can filter
files, directories, by name, size, modification time, and more. Check
man find for details.
If there are spaces or more ‘weird’ characters in the file names, you have to
add -print0 to the end of the find command and use xargs -0 to read
the \0 separated arguments.
You can also replace basic bash loops with xargs:
# Investigate!
for i in {1..10}; do echo $i; done
# here xargs replaces the {} with the line from stdin
# and when there are more lines, it runs the command multiple times
seq 1 10 | xargs -I{} echo {}
# here xargs appends all the lines from stdin to the command
seq 1 10 | xargs echo
What have we learned?
use
findto list files in a directory treeuse
xargsto convert stdin to command line argumentsif your filenames contain spaces, use
-print0and-0to use\0as separatoruse
-I{}to run the command multiple times with different argumentsuse
seqto replace bash loops
Scale up to multiple cores
parallel is a substitute to xargs. The primary difference is that by
default parallel runs one instance of the command per each CPU core on your
machine. Modern machines do have four and more CPU cores.
Additional parallel has a ‘nicer’ and more powerful syntax.
Do control the number of jobs (-j) only when sharing the machine with
someone, or when you’re sure that your task is IO bound. Otherwise
parallel does a good job choosing the number of tasks to run for you.
Note
Parallelizing things IS difficult. There’s no discussion about that. There are some rules of thumb, which can help - but if you want to squeeze out the maximum performance from your machine, it’s still a lot of ‘try - monitor performance - try again’ cycles.
To get good performance it is important to know what happens during data processing: First the data is loaded from hard drive to memory, then from memory to the CPU, the CPU does the calculation, then the results have to get to the memory and saved to the hard drive again. Different workloads take different amounts of time in each step.
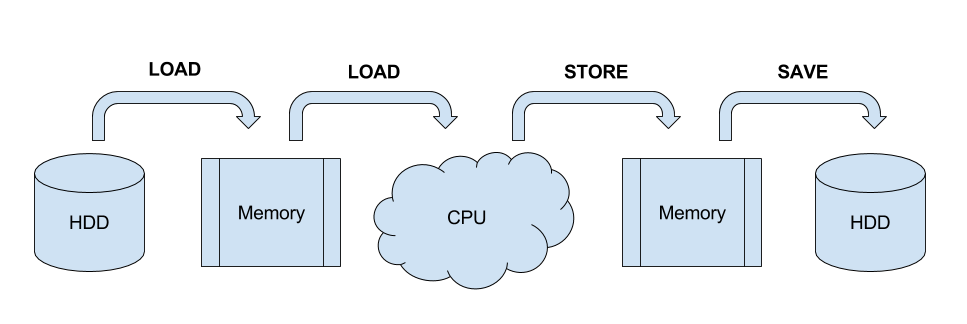
In general, you need a work unit which takes much longer to calculate than
it takes to load the data from the hard drive (compare times of pv data >
/dev/null to pv data | your-task > /dev/null), usually a good work
unit takes on the order of minutes. When disk access seems to be the
limiting factor, you can try to compress the data with some fast compressor
like lz4. Do not parallelize disk intensive tasks, it will make
things only slower! If you still want to use parallel’s syntax, use
parallel -j1 to use only single core.
The most powerful thing about parallel is it’s substitution strings like
{.}, {/}, {#} - check man parallel.
parallel echo Ahoj ::: A B C
parallel --dry-run echo Ahoj ::: A B C
parallel echo File: {} found! ::: data/*.fastq
parallel echo File: {/} found! ::: data/*.fastq
parallel echo File: {/.} found! ::: data/*.fastq
Note
If your data is a single file, but the processing of one line is not
dependent on the other lines, you can use the split command to create
several files each with defined number of lines from the original file.
What have we learned?
use
parallelto run the same command for multiple filesuse
-jto control the number of simultaneous jobsuse
{}to access the input argumentsuse
{.},{/},{#}to access parts of the input arguments